Article by: Christophe Pichavant and Jacques P. Tremblay
Introduction
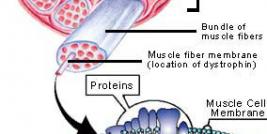
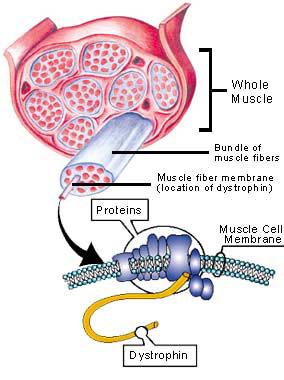
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease characterized by the absence of dystrophin in the muscle. This large protein of 427 kDa is encoded by a 14 kb mRNA (79 exons) (1). This dystrophin protein is located under the membrane of the muscle fiber and interacts with other trans-membrane proteins. It is needed to insure mechanical stress resistance during muscle contraction. The lack of dystrophin weakens the sarcolemma and thus makes fibers less resistant to stress. When a fiber is damaged, the satellite cells (stem cells located near the muscle fibers) are activated and proliferate to allow fiber regeneration (2). In the case of a DMD patient, fibers are frequently damaged due to the lack of dystrophin. The regeneration is also ensured by the proliferation of satellite cells though their number decreases rapidly and when there are no more satellite cells, the average diameter and the number of fibers progressively decrease bringing the development of fibrosis and fat infiltrations into the muscles. There is currently no treatment for DMD and life expectancy is between 20 to 25 years of age.
In 1991, our laboratory obtained evidence of the feasibility of a cell transplantation to restore dystrophin in a DMD patient using an histocompatible donor (3). More recently, in a Phase I clinical trial and in a compassionate treatment, we have been able to restore the expression of dystrophin in up to 34% of the muscle fibers of DMD patients (4, 5). This clinical trial used immuno-incompatible donors with the patient, therefore the patient were immunosuppressed to avoid the rejection of the transplanted cells. Nevertheless, the use of immunosuppressive drugs may induce several adverse effects such as increased risks of cancer, increased risks of infections, nephrotoxicity, neurotoxicity, etc.
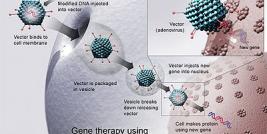
One way to eventually avoid the immune problems associated with allogeneic cell transplantation is to transplant autologous cells which have been genetically corrected. This technique has been proven effective with a truncated version of dystrophin in the mouse and in the primate models (6). This technique is called “ex vivo gene therapy”. Another approach, to avoid these problems, is to directly electrotransfer a cDNA coding for the gene of interest into muscles.
Mouse model of DMD
Although mice mutated with the dystrophin gene do exist, there is currently no mouse model, which has all the clinical characteristics of a DMD patient. These mice called “mdx” for X chromosome-linked muscular dystrophic are mice resulting from a spontaneous mutation in inbred mice. This is a nonsense mutation at base 3185 of the mRNA that causes a premature stop codon within the exon 23 (7). Even if this mutation leads to an absence of the dystrophin in muscles, the development of fibrosis and fat infiltrations in mouse muscles is less important than in DMD muscles (8). Nevertheless, the mdx mouse is a good model and is the most used mouse model to study DMD.
Gene therapy history
Since the first clinical trial of gene therapy in 1990 (9), there was a big interest for this therapeutic approach. However in 1999, a major setback occurred with the death of a patient treated with an adenovirus for OTCD. This death is believed to have been triggered by a severe immune response to the adenovirus carrier (10). In 2002, another issue occurred in a clinical trial for SCID with the use of a retrovirus: one of the 10 patients treated developed T-cell leukaemia (11).
Since that time, the scientific community is more cautious with the use of virus on humans. Nevertheless, laboratories continue to think that gene therapy can be an interesting approach to cure many diseases. In the DMD context, several groups obtained promising results with the gene therapy that are described below.
Non viral gene therapy in DMD
As already mentioned, the size of the cDNA of the dystrophin is large, which brings certain complexity to introduce this plasmid in muscular fibers. There are truncated forms of the dystrophin, the mini of 6 kb and the micro of 4 kb approximately; though only the mini-dystrophin version is completely functional (12). The muscle fibers have a long lifespan since they are in a post-mitotic stage, this allows the expression of a plasmid introduced into muscle to be prolonged. Indeed, a simple intramuscular injection of plasmid coding for the dystrophin can be effective and in 2004, a phase I clinical trial realized on 9 patients DMD or BMD (Becker muscular dystrophy) was realised. Up to 6% of dystrophin positive fibers were observed on one patient after the injection of the plasmid coding for dystrophin (13). This study shows that a simple injection of plasmid is not enough to induce a high restoration of dystrophin. Indeed, plasmid needs to be electrotransferred in muscles to obtain a good percentage of dystrophin positive fibers (14, 15). The electrotransfer is a non-viral method, which allows through an electric field to diffuse and to introduce a plasmid previously injected into the muscle.
Another non-viral method, the PhiC31 system enables the insertion of a transgene used in an adenovirus or with nucleofection. Indeed, the association “PhiC31 + nucleofection” has allowed to introduce the full length dystrophin in MPCs (16) and to obtain its expression in muscles after their transplantation (unpublished results). However, this PhiC31 system must be improved to obtain a better efficiency and specificity of integration. Another non-viral gene therapy is based on exon-skipping, it eliminates one or several exons to restore the reading frame of the dystrophin mRNA and thus its expression. In that case, it is an oligonucleotide anti-sense that is used. Interesting results were published [17, 18, 19) but since there are many different deletions or mutations which cause DMD, different oligonucleotides will have to be used for different groups DMD patients. Furthermore, certain patients can not be treated by this technique, for example those who have a large deletion. There are also controversies about the stability and the toxicity of oligonucleotides used to perform the therapeutic exon-skipping.
Viral gene therapy in DMD
Retroviruses can not be used to treat DMD because they are incapable to transduce quiescent cells, which make up the majority of muscle cells. A therapy with herpesvirus is also not possible because of a poor infection of the muscle cells in vivo due to the large size of the virus (20, 21).
Since the development of the adenovirus’ third generation, there was a strong enthusiasm for this vector. Full length dystrophin but also utrophin, a homologous protein to dystrophin, which binds to the dystrophin complex, were used. Good results, i.e., 50% dystrophin (22) and 95% utrophin (23) positive fibers were observed after the injection of adenovirus directly in the mouse muscles. Nevertheless, with this type of vector, two problems remain: the first one is the absence of integration even if experiments show a stability of the transgene during at least one year (24). The second is its immunogenicity because many patients already have circulating antibodies against adenoviruses. Furthermore, if a second injection of this vector is made, it might induce a fatal immune reaction except if the patient is under a sustained immunosuppressive therapy.
The main advantages of lentivirus vectors are that they are integrative and that they can transduce quiescent cells (25). Thus the infected satellite cells could ensure a sustained expression of the transgene despite many cycles of muscle degeneration/regeneration. However, the encapsidation capacity of lentivirus only allows the inclusion of micro-dystrophin. With lentivirus vectors, the technique called “ex vivo gene therapy” has already shown its efficiency with the transplantation of genetically modified human and monkey MPCs in immunodeficient mice and in monkeys (6). Cossu’s laboratory has also shown a good expression of dystrophin after intra-arterial delivery of genetically modified mesoangioblasts into dystrophic dogs but the dogs had to be immunosuppressed. The systemic delivery of mesoangioblasts may however have important side-effects with the accumulation of cells in different tissues such as the lung (26).
A last vector studied to treat the DMD is the Adeno-associated virus (AAV), its encapsidation capacity does not allow to contain the full length dystrophin but only to carry its truncated micro-dytrophin versions. A strong restoration of dystrophin was observed in dystrophic dogs after intramuscular, regional limb delivery approach or intravenously delivery of AAV carrying the dog micro-dystrophin (27, 28). However the dogs had to be immunosuppressed to prevent an immune reaction against the systemically delivered vector. Another difficulty is to obtain the necessary quantity of virus to treat a patient. An additional characteristic of this vector is its capacity of persistence in muscle which is important but not definitive contrary to wild type AAV.
Ex vivo gene therapy in DMD
Ex vivo gene therapy consists of genetically modifying cells before their transplantation in muscles. There are different parameters to define for this gene therapy. The first one is the time of expression of the transgene in the cells.
To obtain a permanent expression of the transgene it must be integrated. The lentivirus is a good vector to transduce and integrate small transgenes in cells, as already mentioned. The integration of bigger transgenes, it is more problematical because adenovirus and herpesvirus are not integrative. Nevertheless, positive results were obtained with both vectors in ex vivo gene therapy for DMD but the expression of dystrophin in muscles grafted with the genetically modified cells was low (29). Modified adenoviral vectors including the AAV.ITRs and Ad serotype 50 fibers, permit a Rep78 dependant integration and human MPC infection. A group has demonstrated that their modified vector can induce the expression of dystrophin in transduced human MPCs (30) but no in vivo result has been published with this vector.
For transient expression of a large transgene, it can be transducted with an adenovirus but no positive result has been published with the graft of genetically modified MPCs with an adenovirus carrying the full length dystrophin. For smaller transgenes, non-integrating lentiviral vector allows an efficient gene transfer in MPCs (31). However, if the transgene is really small, a simple transfection can be used. In ex vivo gene therapy in DMD, there is no interest for a transient dystrophin expression. Transient expression of a transgene in MPCs may however be useful to improve the viability and the migration of the grafted cells.
Conclusion
There are different methods to introduce the dystrophin cDNA or its truncated versions in muscles. The best method to obtain micro-dystrophin expression is its direct delivery in muscles with the AAV vector. This method has proven its efficiency but an immunosuppressive treatment may be necessary to obtain a long-term expression of micro-dystrophin in the injected muscles. An approach to avoid the immune problems associated with direct viral delivery is to genetically modify MPCs before their transplantation into muscles. The lentiviral vector allowed to obtain the best results with this ex vivo method. In spite of a weaker expression of dystrophin with this method, it allows to genetically modify a patient’s own cells before re-transplanting them without immunosuppression. Electrotransfer also allows to introduce the truncated and the full length versions of dystrophin into muscles. However, the efficiency of this technique is lower especially with the full length dystrophin cDNA than the genetic modification of MPCs with lentiviral vector.
To accept the direct injection of virus into muscles as a potential treatment of DMD patients, the absence of immune response has to be demonstrated or controlled; and for ex vivo gene therapy, its efficacy has to be improved.
About the Authors
Christophe Pichavant is a graduate student working in the lab of Jacques P. Tremblay at the Unité de Recherche en Génétique Humaine, Centre de recherche de CHUL, CHUQ, Faculté de Médecine, Université Laval, Sainte-Foy, Québec, Canada