Dr. Tremblay’s laboratory contributes to the development of treatments for Duchenne muscular dystrophy (DMD). We have an extensive expertise in cell transplantation, histology, immunohistology, immunology and gene therapy. We principally focus on the transplantation of normal or genetically modified Muscle Precursor Cells (MPCs) into muscles to restore the dystrophin.
![]()
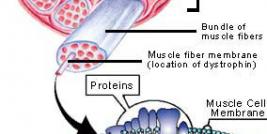
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease characterized by the absence of dystrophin in the muscle. This large protein of 427 kDa is encoded by a 14 kb mRNA. This dystrophin protein is located under the membrane of the muscle fiber and interacts with other trans-membrane proteins. It is needed to insure mechanical stress resistance during muscle contraction. The lack of dystrophin weakens the sarcolemma and thus makes fibers less resistant to stress. When a fiber is damaged, the satellite cells (stem cells located near the muscle fibers) are activated and proliferate to allow fiber regeneration. In the case of a DMD patient, fibers are frequently damaged due to the lack of dystrophin. The regeneration is also ensured by the proliferation of satellite cells though their number decreases rapidly and when there is no more satellite cell, the average diameter of fibers progressively decrease bringing the development of fibrosis and fat infiltrations into the muscles. There is currently no treatment for DMD and death occurs generally between 20 to 25 years of life.
In 1991, our laboratory demonstrated the feasibility of a cell transplantation to restore dystrophin in a DMD patient. Since that time, we have worked our way up to improve the technique and to obtain more successful transplantations. Though, as discussed below, three main problems remain with the clinical use of cell transplantation:
The specific rejection of the grafted cells. In transplantation, donor cells must be immuno-compatible with the donor, otherwise cells will be rejected. In clinical trial context, MPCs were often from a donor immuno-incompatible with the patient, thus the patient must be immunosuppressed to avoid the rejection of the transplanted cells. Our laboratory has obtained good results with the use of Tacrolimus in mouse, primate models and in DMD patients [1]. Nevertheless, the use of immunosuppressive drugs may induce several adverse effects such as increased risks of cancer, increased risks of infections, nephrotoxicity, neurotoxicity, etc. An alternative approach to eventually avoid the immune problems associated with allogeneic cell transplantation is to induce tolerance to the donor antigens. We have demonstrated that a treosulfan treatment combined with single cyclophosphamide dose and a donor bone marrow transplantation permit to develop donor-specific immunological tolerance and to obtain dystrophin expression in dystrophic mice after allogeneic cell transplantation [2]. Another possibility is to transplant autologous MPCs (from the DMD patient himself), which have been genetically modified to carry the normal dystrophin cDNA. This technique has been proven effective with a truncated version of dystrophin in the mouse and in the primate models [3].
The early death of the transplanted cells. Using TUNEL, Y chromosome PCR and [14C] thymidine methods, our laboratory has demonstrated that about 70 % of cells die (by necrosis and apoptosis) during the three days following their transplantation. There are several mechanisms responsible for this high mortality. The first one is inflammation: indeed, one hour following cell transplantation, neutrophils are already located on the graft site, followed by macrophages [4]. Several factors secreted by these cells such as interleukins, cytokines and caspases can induce apoptosis. Another mechanism implicated in the death of the transplanted cells is anoïkis (apoptosis triggered by the loss of cell anchorage). Modulation of apoptotic factors (Bit1, Bcl-2 and FADD) reduced this apoptosis [5]. Our group also demonstrated that over-expression of VEGF improved survival and engraftment of MPCs (myoblasts) in mouse muscles, thus the hypoxia/isthemia is also implicated in this myoblast death [6]. A last mechanism responsible for early myoblast death is the injection technique itself: cells are exposed to various physical stresses during their preparation and injection.
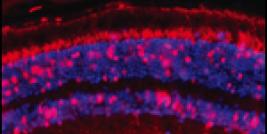
The low migration of transplanted cells. Following the transplantation of normal MPCs, dystrophin is only observed near the injection trajectories, indicating that the grafted cells do not migrate far from the injection trajectories. Our laboratory has demonstrated that several approaches (e.g., treatments with bFGF or IL-4) able to improve myoblast migration capacity in vitro or in mouse muscles but did not permit to reduce the number of injections in a large animal model. One of the main problems of myoblast migration is to induce chemo-attraction of the grafted MPCs towards the host muscle fibers so that they fuse with them.
Our laboratory is also interested by the myostatin pathway. We have indeed increased the MPC transplantation success by blocking myostatin with follistatin or by transplanting MPCs with a non functioning myostatin receptor [7].
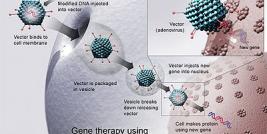
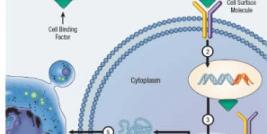
We are also involved in gene transfer. Indeed several of our results were obtained by genetically modifying cells. Gene therapy allows (1) to overexpress a gene already present in the cell: the delivery (before cell transplantation) of a plasmid coding for VEGF into muscles improved the MPC survival and the graft success, (2) to add a gene such as the dystrophin cDNA in DMD cells [3] or (3) to reduce the expression of a gene: transfection of MPCs with a siRNA against Bit1 improved MPC survival [5].
Adenovirus, Ad/AAV, Lentivirus and Retrovirus are used in our laboratory to transduce cells, but non-viral methods (plasmid coding for one or several transgenes) are also used. Dystrophin expression was observed, in muscles, after the graft of MPCs nucleofected with a plasmid coding for dystrophin full length cDNA (>14kb). We have also used electrotransfer to introduce a dystrophin plasmid directly into muscle fibers in vivo. Hypertrophied fibers were also observed following the electrotransfer of a plasmid coding for the follistatin cDNA, this hypertrophy increased the resistance of the muscle fibers to excentric contractions [7]. We have thus obtained very good results by using viral and non-viral gene therapies.
![]()
All these improvements have allowed us to do a phase I clinical trial of myoblast transplantation in 10 DMD patients. Following the transplantation of normal allogeneic myoblasts in these patients using tacrolimus immuno-suppression, dystrophin was expressed in up to 34% of in muscle fibers [1]. Thus MPC transplantation is an interesting therapeutic approach for the treatment of DMD patients.
Recent Publications
1. Skuk, D., et al., First test of a "high-density injection" protocol for myogenic cell transplantation throughout large volumes of muscles in a Duchenne muscular dystrophy patient: eighteen months follow-up. Neuromuscul Disord, 2007. 17(1): p. 38-46.
2. Stephan, L., et al., Induction of tolerance across fully mismatched barriers by a nonmyeloablative treatment excluding antibodies or irradiation use. Cell Transplant, 2006. 15(8-9): p. 835-46.
3. Quenneville, S.P., et al., Autologous transplantation of muscle precursor cells modified with a lentivirus for muscular dystrophy: human cells and primate models. Mol Ther, 2007. 15(2): p. 431-8.
4. Skuk, D., et al., Resetting the problem of cell death following muscle-derived cell transplantation: detection, dynamics and mechanisms. J Neuropathol Exp Neurol, 2003. 62(9): p. 951-67.
5. Bouchentouf, M., et al., Induction of Anoikis following myoblast transplantation into SCID mouse muscles requires the Bit1 and FADD pathways. Am J Transplant, 2007. 7(6): p. 1491-505.
6. Bouchentouf, M., et al., Vascular endothelial growth factor reduced hypoxia-induced death of human myoblasts and improved their engraftment in mouse muscles. Gene Ther, 2008. 15(6): p. 404-14.
7. Benabdallah, B.F., et al., Inhibiting myostatin with follistatin improves the success of myoblast transplantation in dystrophic mice. Cell Transplant, 2008. 17(3): p. 337-50.